医疗侦探小组selenoprotein1 / EPT1
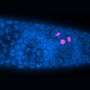
, 1 year (C-D) and 4 years (E-F) and show an absence of myelin where expected (arrows) and increasing atrophy (asterisks). Researchers traced the myelin deficiency to EPT1-induced shortages of the phospholipid plasmalogen, a downstream product made from phosphatidylethanolamine. Previously, EPT1 was not known to be essential for plasmalogen biosynthesis. Credit: Ayelet Eran and coauthors")
婴儿看起来健康。但在两个小时内,他有严重的癫痫发作。
医院工作人员在海法Rambam医学中心,以色列,在执行例行检查时发现婴儿不适。他们把他转到新生儿重症监护室马上,开始进行测试,希望能达成一个诊断。但每个测试呈阴性。他的结果看起来正常。
这个问题很简单:这孩子怎么了?
需要数年才能找出答案。Rambam医学中心的研究人员,日本RIKEN大脑科学研究所,和其他机构在这两个国家宣布他们解决医学谜在6月刊的一篇文章脂质研究期刊》的研究。
一个神秘的遗传疾病
代谢性疾病专家,汉娜Mandel博士的团队评估了婴儿在NICU早在2011年。癫痫在新生儿可能来自于各种各样的导致一些比其他的更常见。什么在这个婴儿的一般外观为他发作的原因提供了线索。十多个常规检测回来正常。孩子没有感染,没有问题与已知代谢物,在他的尿或脑脊液没有线索。在完成一系列常规检测以及额外的代谢检查,曼德尔说,他们什么也没找到,可以解释孩子的疾病。
医疗队送孩子,这时候三周大,大脑扫描。图像显示缺乏髓,必要的脂肪层周围神经。髓鞘就像周围的塑料绝缘电线。如果绝缘没有,线的这种情况下,神经元无法携带电信号或他们应该一样快。他们认为,这一缺陷可能影响孩子的发展。
随着年龄的增长,孩子错过了里程碑,从未在自己的移动。后续的扫描显示,他的髓磷脂是仍然没有发展,他的大脑和其他地区开始萎缩。
孩子4岁时,奥利Elpeleg博士专家罕见遗传疾病在耶路撒冷hebrew大学医学中心,领导一个团队,男孩的基因组测序和分析,寻找基因突变,他可能已经继承了他的父母,谁是堂兄妹。
Elpeleg确认13网站在男孩的基因组中,他继承了非常罕见的编码从父母双亲那里都遗传变异。通过检查他的健康的家庭成员的基因,这些网站的团队可以排除许多引起孩子的问题。负责疾病影响的突变研究人员发现一个叫做EPT1基因。与健康的控制相比,病人的信使rna编码EPT1蛋白异常缩短。这是第一次一个病人被诊断为EPT1突变。
遗传学家发现了突变时,曼德尔直接去PubMed数据库住房医疗和生物研究。她看,她说,“对于那些有兴趣学习EPT1的致病性突变。”
她发现世界专家在基因上的问题。
建立一个国际合作
几年前,Yasuhiro Horibata Yoshio Hirabayashi的实验室,博士后研究脂质理研大脑科学研究所生理学在著名的日本。
两人发现EPT1基因编码的一种酶,这种酶把收尾工作生产的脂质称为磷脂酰乙醇胺,或简称为PE。PE约占所有大脑中的磷脂的五分之一。他们发表了他们的研究结果在2007年的一篇论文脂质研究期刊》的研究。
曼德尔发现,纸和Hirabayashi写道。很快,她,Hirabayashi Horibata,那时Dokkyo大学助理教授,孵化合作通过电子邮件,和以色列的团队组织样本从子运往日本。
日本团队测量酶活性在孩子的细胞,并证实它很低而健康的控制,因为错误地缩短蛋白是立即被细胞的质量控制系统。他们寻找孩子的皮肤细胞的变化,测量所有的脂肪细胞产生。令他们吃惊的是,缺乏EPT1稍微减少了孩子的体育的组织。这是意想不到的,因为体育是主要的已知EPT1活动的产物。
然而,EPT1短缺,极大地降低了另一个分子的数量,缩醛磷脂,脂质产品由PE在髓磷脂丰富。看来孩子的细胞能够雇佣其他酶来弥补PE生产;然而这并没有救下缩醛磷脂合成。研究人员得出结论,尽管其PE-manufacturing工作由另一个酶的作用,重复EPT1使髓磷脂在正常大脑至关重要。这个工作团队中描述的新捷豹路虎。
对未来的希望
研究人员解决了遗传的男孩的困惑背后的神秘疾病。去年英国团队报道患者相似的症状和类似的突变,使以色列男孩第二EPT1障碍患者在医学文献报告。
很多人住在一起罕见的遗传疾病男孩的治疗选择是有限的。今天,病人半身不遂,失明、失聪,和他的癫痫已经恶化。
然而,曼德尔强调的积极方面孩子的生活。“他住在一个美丽的阿拉伯村庄在以色列北部,在一个美丽的房子,有他的父母和他的妹妹,”她说。“白天,他参加一个康复中心他家附近的村庄,在那里他获得一个能想到的所有的辅助医疗的支持。”
与此同时,医生给男孩的父母遗传咨询基于他们的研究成果。植入前胚胎遗传学诊断”这对夫妇的目标是为将来怀孕,“曼德尔说。
更多信息:Yasuhiro Horibata et al,乙醇胺磷酸转移酶1(硒蛋白I)是至关重要的缩醛磷脂在人类的神经系统开发和维护,脂质研究期刊》的研究(2018)。DOI: 10.1194 / jlr.P081620