研究人员称量方法更准确地测量基因组测序
在2003年宣布的人类基因组被测序的欣喜中迷失了一个基本问题:我们如何确定个人的基因组已正确阅读?
虽然第一个完整的,单个基因组是十年前测序的,但鉴于广阔遗传变异在全球70亿人中,更不用说在近亲之间的化妆差异,个人对个人进行准确测序的问题继续困扰研究人员。
现在,公司预计他们可以以1,000美元的价格对基因组进行排序,从几年前的25,000美元下降,并为开发“个性化”药物的努力,此事在当今市场上的重要性提高了。这些更便宜的努力依赖于新技术,这些技术假设科学家可以继续使用标准的shot弹枪方法将基因组随机切碎成较小的碎片,然后将其重新组装为算法。具体而言,今天的较低成本是通过将DNA分解成更细小的碎片,并迅速且廉价地阅读其中的大量成本来实现。但是,尚不清楚如何评估较新的组装算法的准确性和基本的shot弹枪方法,尤其是在较早的准确性的情况下基因组数据值得怀疑。
确认个人基因组测序准确性的特定挑战之一就是将一个人的表型或身体特征与他或她相匹配基因型, 或者基因组成。特别是,这是成功开发个性化药物的障碍,这是在第一次测序后不久预测的人类基因组,但尚未真正实现。
在期刊的一篇文章中PLOS ONE,纽约大学库兰特学院的研究人员数学科学评估一些当前的方法来对单个基因组进行测序,该研究是对这些实践功效的“应力测试”。
研究人员采用了旨在识别基因组的关键或代表性特征的测试程序,以及这些特征中的每个特征与其他功能如何相关。
“大多数当前的技术在组装基因组时,在遇到重复区域时会犯一些错误 - 在其中构成字母的子弦脱氧核糖核酸计算机科学和数学教授和研究的高级作者Bud Mishra解释说,链条在基因组中的许多地方重新发生。。”
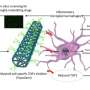
为了测试这些程序的可行性,纽约大学研究人员依靠由基因组学家和生物信息学家的公共财团开发的开源软件AMOS的功能集合。如果一种方法对个人的整个基因组进行了准确测序,研究人员假设了该方法的创建组成部分,则应“合并”,并将与其他辅助数据一致,例如“ Mate Pairs”,“光学图”,“光学图”或“ strobed”序列,“所有这些构成了基因组中的远程信息。当前,序列对的使用在序列组装和验证算法中很常见,但不是其他两个。
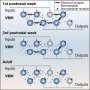
尽管他们发现所有检查的方法对个人基因组进行测序的缺点,但一些汇编者表现出了希望。纽约大学研究人员的结论源自称为特征反应曲线(FRCURVE)的程序,该程序有效地显示了不同组装者如何能够处理大型复杂基因组中不同区域和不同结构的全球图。通过这种方式,它还指出了一个汇编商如何以牺牲另一种质量的方式进行交易。例如,它显示了一个基因组组装者可能试图将一组基因集中到基因组的连续部分中的积极性,同时错误地重新排列了其正确的顺序和拷贝数。
“此类错误具有重要的后果,尤其是在使用该技术来研究肿瘤的基因组时,通常可能是高度异质的,使每个肿瘤细胞的细胞都变得基因组重新排列和突变与邻居的变化截然不同,” Mishra解释说。